Post-synthetic modification of metal-organic frameworks: a rational route to structural and chemical diversity
In the realm of nanoporous materials, metal-organic frameworks (MOFs) stand apart from zeolites, their inorganic counterparts, by virtue of their diverse structures and functionalities, the result of having a virtually limitless library of organic ligands as their building blocks. However, the structural diversity and conformational flexibility of these constituent ligands, coupled with their weaker coordination bonds, leads to a lack of predictability in the MOF structures obtained (Scheme 1, A). Hence, while one of the most touted features of MOFs is the ability to tailor their pore cavity environment and topology, there are very few examples in which the organic linker can be changed systematically whilst maintaining the same framework structure or, conversely, where the same ligand can be used to construct MOFs with several different structures. My research in the Hupp/Nguyen groups involves developing strategies for rationally achieving both of these elusive goals via post-synthesis modification.
|
A) Traditional MOF Synthesis |
B) Synthesis of structural isomers |
C) Introduction of chemical diversity |
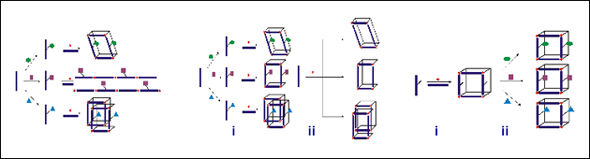 Scheme 1: Traditional MOF synthesis (A). Route to structural isomers (B), and introducing chemical diversity (C) via post-synthesis chemistry.
Scheme 1: Traditional MOF synthesis (A). Route to structural isomers (B), and introducing chemical diversity (C) via post-synthesis chemistry.
Ligand-elaboration as a strategy for engendering structural diversity in porous metal-organic framework compounds
This work demonstrates our ability to tailor the chemical pore environment of MOFs and, at the same time, to engender structural diversity in these materials via organic synthesis (Scheme 1, Bi). A series of Zn-based, mixed-ligand metal-organic frameworks were constructed, in which the connectivity — and more exceptionally, the level of interpenetration of the structures — is influenced by the systematic alteration of the organic ligand remote from the coordination site (Figure 1). Small changes in the side groups of the ethynylene dibenzoate struts lead to three different structures, all with large pores and different gas adsorption behaviors. To our knowledge, this is the first example in which such ligand-influence assembly has been demonstrated systematically, and these results give credence to our hypothesis that we can rationally synthesize MOF structural isomers using strategy (B, Scheme 1).
 Ligands
Ligands
Network units. Zinc: yellow, carbon: grey, oxygen: red, nitrogen: blue, bromine: brownSchematic representations of the network interpenetration for each MOF.
Schematic representations of the network interpenetration for each MOF.
|
Figure 1: Three different network topologies observed using very similar ligands. The H substituents gives a four-fold interpenetrating, “diamond-like” structure, while the lager substituents give paddle-wheel frameworks. The similarly sized Me and Br substituents give identical, three-fold interpenetrating, topology, while the CH2OH substituents results in a more open, two-fold interpenetrating framework. |
Covalent surface modification of a metal-organic framework: selective surface engineering via CuI-catalyzed Huisgen cycloaddition
The ligands described in Section 2 can also be used to demonstrate a strategy for achieving structure-independent chemical diversity in MOFs (Scheme 1C). The assembled MOF materials bearing trialkylsilyl-protected alkynes can be post-synthetically deprotected to give MOF crystals decorated with terminal acetylenes (Figure 3). These MOFs are then ready for elaboration with a variety of organic azides via CuI-catalyzed [3+2] cycloadditions. We modified our ethynyl-terminated MOF surface with two different organic azides. As a proof-of-concept, the fluorophore ethidium bromide monoazide was attached to the surface of our MOF, resulting in a porous solid that fluoresces under visible light excitation. We then extended this strategy to reverse the hydrophobicity of our material by attaching polyethylene glycol chains to its surface (Figure 3). This work highlights a powerful approach to realizing one of the “holy grails” of MOF chemistry: the fine-tunability of MOF properties through rational design and synthesis. Current efforts are directed towards assembling noninterpenetrated MOFs, with larger pores, so that the elaboration can be carried out throughout the interior of the crystals.
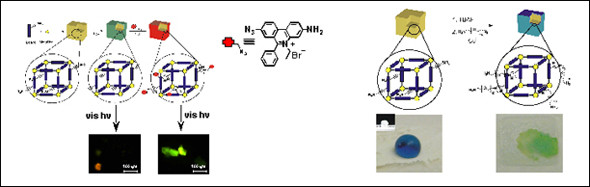 Figure 3
Figure 3